¿Que sucede en nuestra región?
¿Que sucede en nuestra región?
1 de julio de 2020.
En Argentina actualmente existen 29 secuencias de SARS-CoV-2. A continuación se muestra una figura donde a la izquierda se encuentra un árbol filogenético construido a partir del alineamiento múltiple de 375 secuencias aisladas de pacientes de países de latinoamérica (GISAID), incluyendo las secuencias argentinas, donde a su vez se indica el linaje a la cual corresponde cada secuencia. A la derecha se indica la procedencia geográfica donde fue obtenida cada una de las secuencias.
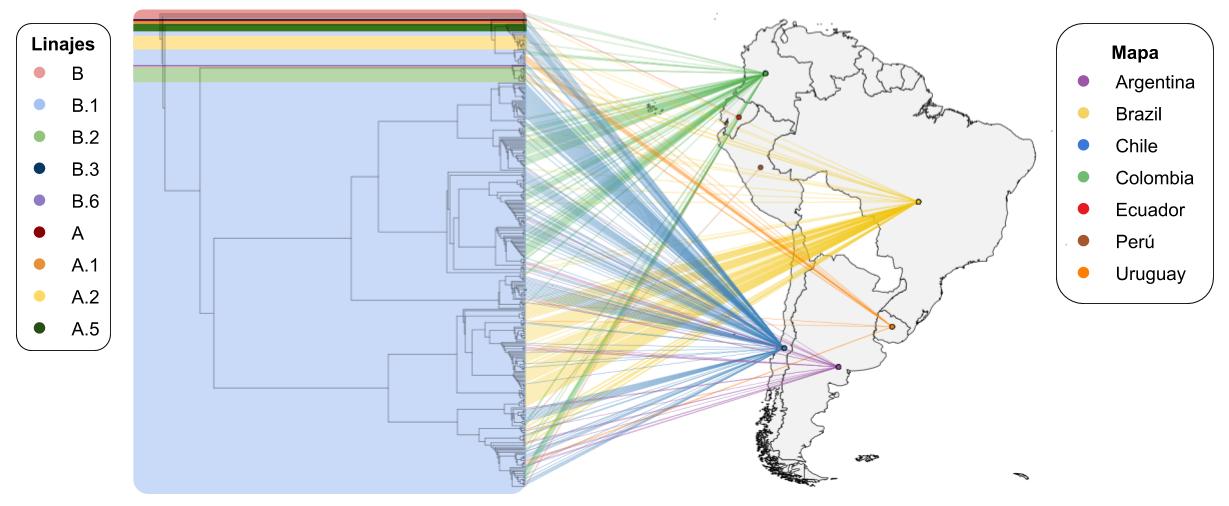
Figura 1. Árbol Filogenético de SARS-CoV-2 en Sudamérica. Las secuencias y coordenadas geográficas fueron obtenidas a partir de la metadata disponible en la base de datos GISAID a la fecha 12 de Junio, 2020. El árbol filogenético fue creado con IQtree e integrado con el mapa a partir la librería de R, phytools. Cada secuencia está conectada por una línea coloreada con su país de procedencia (Referencia, cuadro izquierdo Mapa). Se analizó el linaje de las secuencias utilizando el programa Pangolin y se las coloreó sobre las ramas del árbol filogenético (Referencia, cuadro derecho Linajes).
Observando la Figura 1 encontramos agrupación de secuencias de ramas de la misma procedencia, indicando que las secuencias de una región poseen mayor similitud respecto al resto.
A los fines de facilitar el entendimiento de los parentescos filogenéticos de las secuencias se establecen sistemas de clasificación por linajes. En esencia, a partir de un análisis filogenético se asignan nombres a ciertas ramas. De este modo, al decir que una secuencia forma parte de un determinado linaje, es posible entender su ubicación en el árbol rápidamente y con qué otras secuencias está más emparentada.
Una manera de asignar linaje a una secuencia en particular de manera rápida es mediante el programa Pangolin. Este programa toma una secuencia a analizar, la incluye en un alineamiento de secuencias representativas de todos los linajes preestablecidos [1] y luego computa una filogenia utilizando un árbol inicial guía que es representativo de la filogenia de los linajes. Una vez obtenido un árbol definitivo, Pangolin determina el linaje de la secuencia en función de su ubicación en el árbol y del linaje al que pertenecen sus vecinos más próximos.
Al completar el análisis, Pangolin devuelve una tabla donde cada fila es una de las secuencias analizadas y las columnas corresponden al nombre del archivo donde estaba la secuencia, el nombre de la secuencia, el linaje asignado y los valores de soporte de ramas de Bootstrap [2] y SH-aLRT [3]. Estos dos últimos son estadísticos indicativos de la “credibilidad” de una rama del árbol.
En la Figura 5 se puede observar qué linaje le fue asignado a cada una de las secuencias del árbol y de esta manera, teniendo en cuenta la temporalidad de la pandemia, nos permite hacer inferencias acerca del orígen de los diferentes aislamientos. Por ejemplo, las secuencias argentinas fueron asignadas todas al linaje B.1, que es uno de los principales causantes de los brotes en Europa y América del Norte (https://github.com/hCoV-2019/lineages/). En el caso de las secuencias chilenas, en cambio, podemos observar asignaciones a distintos linajes, incluidos algunos linajes “A.X” (circulantes en Reino Unido, EE.UU, España o Australia). Se pueden observar hasta 8 clusters de secuencias chilenas asignados a diferentes linajes, lo que podría indicar hasta 8 introducciones diferentes del virus generando clusters independientes de circulación local. No obstante, es de considerar que existen otras posibilidades para el caso chileno, como que hayan habido varias introducciones más del virus que no se puedan ver en la filogenia por falta de secuencias. También es importante tener en cuenta que la correlación entre países y linajes no se puede inferir de manera directa. Usualmente un mismo país presenta más de un linaje circulando, aunque suele haber un linaje preponderante.
Referencia
- [1]. Rambaut A, Holmes EC, Hill V, O’Toole Á, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. doi:10.1101/2020.04.17.046086
- [2]. Holmes S. Bootstrapping Phylogenetic Trees: Theory and Methods. Statistical Science. 2003. pp. 241–255. doi:10.1214/ss/1063994979
- [3]. Anisimova M, Gascuel O. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Systematic Biology. 2006. pp. 539–552. doi:10.1080/10635150600755453